Lendületes kutatók vezetésével derítették fel egy ritka, súlyos betegség okát
A nemzetközi kutatócsoport több mint egy évtizeden át tartó genetikai nyomozással jutott el egy család férfi tagjait három generáció óta sújtó kórkép eredetéhez. Eredményeik a betegség kezelése mellett annak megértésében is sokat segíthetnek, hogy miként kezelik sejtjeink az örökítőanyagban található, genetikai kódon túli információkat.
Nem mindegy, hogy valakin a kor vagy a kór hagyta rajta a nyomát, és annak is más üzenete lehet, ha kiirtunk vagy kiírtunk magunkból valamit. Az ékezetek értelemformáló fontossága magától értetődőnek tűnik mindenki számára, de az kevésbé ismert, hogy az írott nyelv ékezeteinek analógjai léteznek az élőlények genetikai információját rejtő nukleinsavakban is. Az élet kódjának „ékezeteit”, vagyis az örökítőanyagban tárolt minden, a nukleotidsorrenden felüli információt (kémiai vagy térszerkezeti változásokat) epigenetikai módosításoknak nevezik. Ezek a módosítások létfontosságúak minden kicsit is bonyolultabb többsejtű élőlény, így az ember számára is, hiszen nélkülük nem lehetnének különböző típusú sejtjeink úgy, hogy genetikai kódjuk (a DNS nukleotidsorrendje) egyébként azonos.
Az elmúlt évtizedekben leginkább a genetikai információ hosszú távú tárolását végző DNS epigenetikai módosításai álltak a figyelem és a kutatások középpontjában, de az utóbbi években egyre több eredmény bizonyította, hogy másik kulcsfontosságú nukleinsavunkat, az RNS-t érintő „epitranszkriptomikus” módosulatoknak is fontos szerepük van a sejtek megfelelő működésében.
Az MTA-SE Lendület Nephrogenetikai Kutatócsoport vezetésével a Semmelweis Egyetem, az ELTE, valamint a University College London kutatói a napokban megjelent tanulmányukban azt mutatták ki, hogy milyen súlyos következményekkel járhat, ha a sejtek egyik „RNS-ékezetét” készítő enzim meghibásodik a szervezetben.
Az Egyesült Államok Tudományos Akadémiájának hivatalos folyóirata, a Proceedings of the National Academy of Sciences lapjain publikált kutatás kiindulási pontját egy, a Semmelweis Egyetem I. Sz. Gyermekgyógyászati Klinikán Szabó Attila professzor által gondozott, ismeretlen eredetű betegségben szenvedő család adta. A Tory Kálmán (SE I. Sz. Gyermekgyógyászati Klinika) által vezetett MTA-SE Lendület Nephrogenetikai Kutatócsoport az elsősorban a fiúkat érintő vese-, szem-, fül- és bélérintettséggel járó kórkép eredetének nyomába eredt. Előbb azonosították a felelős régiót az X-kromoszómán a párizsi Imagine Intézettel való kollaborációban, majd a Kölni Egyetemen szekvenálva a régiót, megtalálták a kóroki variánst a diszkerin enzimet kódoló DKC1 génben. A diszkerin az egyike azoknak az enzimeknek, amelyek az RNS-molekulák „ékezeteiért” felelnek, és amely nélkül számos pozícióban nem jön létre a pszeudouridinnek nevezett módosulás.
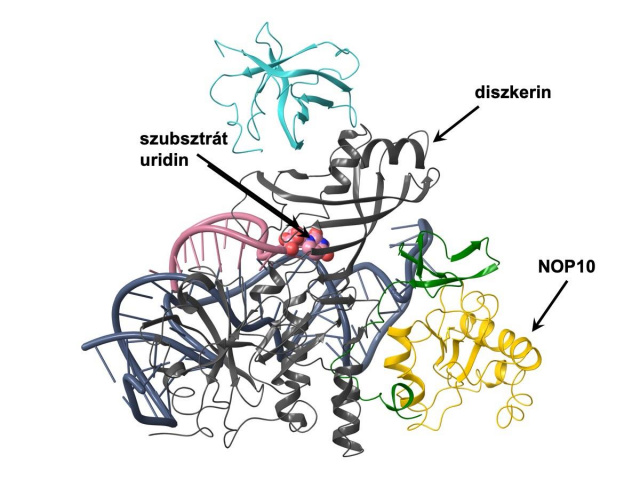 A fehérjekomplex szerkezete, szürkével a diszkerin, sárgával a NOP10 Forrás: MTA-SE Lendület Nephrogenetikai Kutatócsoport
A fehérjekomplex szerkezete, szürkével a diszkerin, sárgával a NOP10 Forrás: MTA-SE Lendület Nephrogenetikai KutatócsoportA Lendület-kutatócsoport rámutatott, hogy a kóros mutáció három generációval korábban, a betegséggel együtt alakult ki a családban. A DKC1 gén szerepe váratlan volt, mert variánsait korábban egy teljesen más, a kromoszómát lezáró telomerek rövidülésével járó betegségben azonosították.
A génhez társuló új betegség szokatlan eredetére a fehérjeszerkezeti kutatások mutattak rá. A diszkerin egy nagyobb molekuláris komplex tagjaként fejti ki a hatását, ahol a különböző fehérjék koordinált együttműködése szükséges ahhoz, hogy a megfelelő RNS-módosítások bekövetkezhessenek.
A Karancsiné Menyhárd Dóra (MTA-ELTE Fehérjemodellező Kutatócsoport) által végzett szerkezeti modellezés alapján az új DKC1-mutáció hatására károsodik a diszkerin kulcsfontosságú katalitikus régiója, és megváltozik a diszkerin kapcsolata egyik partnerével, a NOP10 fehérjével. E számításokat kísérletesen igazolta Balogh Eszter (MTA-SE Lendület Nephrogenetikai Kutatócsoport) és Schay Gusztáv (SE Biofizikai és Sugárbiológiai Intézet) a két fehérje kapcsolatának nyomásperturbációs fluoreszcencia-spektroszkópia vizsgálatával.
A végső bizonyítékot, hogy az újonnan leírt betegséget az enzim károsodása, nem pedig a telomerrövidülés okozza, egy állatmodell szolgáltatta. Varga Máté (ELTE TTK Genetikai Tanszék) csoportja több olyan zebrahalvonalat hozott létre a CRISPR/Cas9 genomszerkesztő rendszer használatával, amely az állatok saját diszkerin enzimjét kódoló génben hordozott mutációkat. A mutáns állatok nagyon hasonló szervi érintettséget mutattak, mint a betegek, és vizsgálatukkal ki lehetett mutatni, hogy a betegség számos tünete mögött a sejtosztódás zavara áll. Ez a fehérjeszintézisért felelős riboszómák hibás működésével hozható összefüggésbe, amit a pszeudouridinek hiánya okoz a riboszómákat is létrehozó RNS-molekulákban.
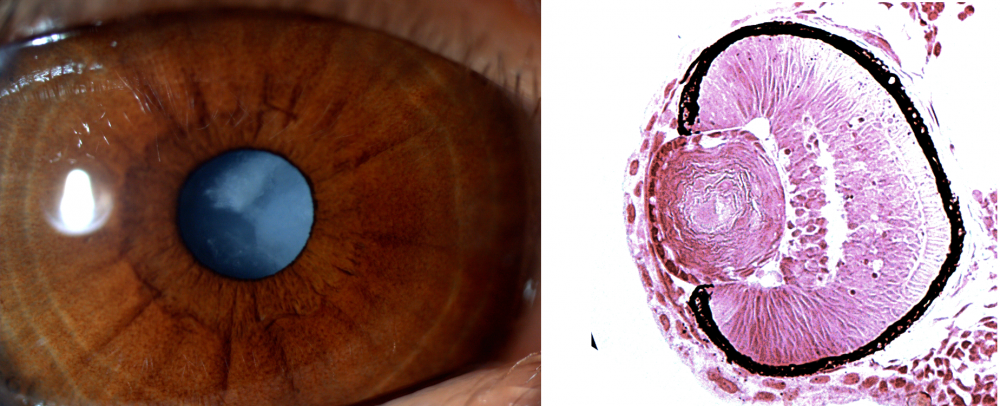 Bal oldalon az egyik beteg szeme, melyen jól megfigyelhető a szürkehályog, jobbra pedig az egyik mutáns zebrahallárva szemének metszete – itt jól látszik, hogy a szemlencse fibrózus szerkezetű (ami szintén a hályog jele) Forrás: MTA-SE Lendület Nephrogenetikai Kutatócsoport
Bal oldalon az egyik beteg szeme, melyen jól megfigyelhető a szürkehályog, jobbra pedig az egyik mutáns zebrahallárva szemének metszete – itt jól látszik, hogy a szemlencse fibrózus szerkezetű (ami szintén a hályog jele) Forrás: MTA-SE Lendület Nephrogenetikai KutatócsoportA több mint egy évtizeden át tartó munka tartogatott még egy meglepetést: két évvel ezelőtt egy londoni kutatócsoport a NOP10 fehérjében is azonosított egy ugyanezen tünetegyüttest okozó variánst, melynek a diszkerin-NOP10 interakcióra gyakorolt hatása megegyezik a diszkerinben talált mutációéval.
A közös közlemény így végül angol, francia, kanadai és német kutatókkal való kollaboráció eredménye lett.
A kutatók így azonosítottak egy új betegségért felelős kórfolyamatot, valamint a diszkerinmediált pszeudouridiláció szerepét a különböző szervek működésében. A Lendület program által finanszírozott kutatás szép példája annak, hogy klinikai megfigyelések hogyan vezethetnek alapkutatásban is fontos folyamatok megismeréséhez, egy betegség okának és mechanizmusának megértéséhez. A kutatásban csak így, több kutatócsoport együttműködésének eredményeképpen kerülhet fel az i-re a pont (vagy vessző).
További információ
Tory Kálmán, MTA-SE Lendület Nephrogenetikai Kutatócsoport
tory.kalman@med.semmelweis-univ.hu
Varga Máté, ELTE TTK Genetikai Tanszék
mvarga@ttk.elte.hu



